Machine learning applications in genetics and genomics
The field of machine learning, which aims to develop computer algorithms that improve with experience, holds promise to enable computers to assist humans in the analysis of large, complex data sets. Here, we provide an overview of machine learning applications for the analysis of genome sequencing data sets, including the annotation of sequence elements and epigenetic, proteomic or metabolomic data. We present considerations and recurrent challenges in the application of supervised, semi-supervised and unsupervised machine learning methods, as well as of generative and discriminative modelling approaches. We provide general guidelines to assist in the selection of these machine learning methods and their practical application for the analysis of genetic and genomic data sets.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
206,07 € per year
only 17,17 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

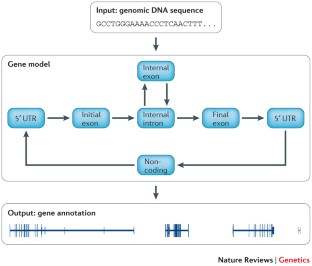
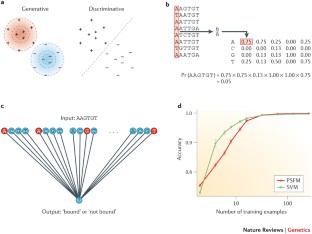
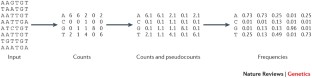
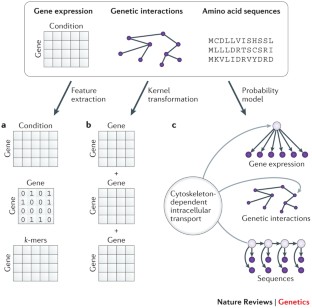
Similar content being viewed by others
Navigating the pitfalls of applying machine learning in genomics
Article 26 November 2021
Decoding disease: from genomes to networks to phenotypes
Article 02 August 2021
Computational analysis of cancer genome sequencing data
Article 08 December 2021
References
- Mitchell, T. Machine Learning (McGraw-Hill, 1997). This book provides a general introduction to machine learning that is suitable for undergraduate or graduate students.Google Scholar
- Ohler, W., Liao, C., Niemann, H. & Rubin, G. M. Computational analysis of core promoters in the Drosophila genome. Genome Biol.3, RESEARCH0087 (2002). ArticlePubMedPubMed CentralGoogle Scholar
- Degroeve, S., Baets, B. D., de Peer, Y. V. & Rouzé, P. Feature subset selection for splice site prediction. Bioinformatics18, S75–S83 (2002). ArticlePubMedGoogle Scholar
- Bucher, P. Weight matrix description of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. J. Mol. Biol.4, 563–578 (1990). ArticleGoogle Scholar
- Heintzman, N. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genet.39, 311–318 (2007). ArticleCASPubMedGoogle Scholar
- Segal, E. et al. A genomic code for nucleosome positioning. Nature44, 772–778 (2006). ArticleGoogle Scholar
- Picardi, E. & Pesole, G. Computational methods for ab initio and comparative gene finding. Methods Mol. Biol.609, 269–284 (2010). ArticleCASPubMedGoogle Scholar
- Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nature Genet.25, 25–29 (2000). ArticleCASPubMedGoogle Scholar
- Fraser, A. G. & Marcotte, E. M. A probabilistic view of gene function. Nature Genet.36, 559–564 (2004). ArticleCASPubMedGoogle Scholar
- Beer, M. A. & Tavazoie, S. Predicting gene expression from sequence. Cell117, 185–198 (2004). ArticleCASPubMedGoogle Scholar
- Karlic, R. R. Chung, H., Lasserre, J., Vlahovicek, K. & Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl Acad. Sci. USA107, 2926–2931 (2010). ArticleCASPubMedGoogle Scholar
- Ouyang, Z., Zhou, Q. & Wong, H. W. ChIP–seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc. Natl Acad. Sci. USA106, 21521–21526 (2009). ArticleCASPubMedGoogle Scholar
- Friedman, N. Inferring cellular networks using probabilistic graphical models. Science303, 799–805 (2004). ArticleCASPubMedGoogle Scholar
- Hastie, T., Tibshirani, R. & Friedman, J. The Elements of Statistical Learning: Data Mining, Inference and Prediction (Springer, 2001). This book provides an overview of machine learning that is suitable for students with a strong background in statistics.BookGoogle Scholar
- Hamelryck, T. Probabilistic models and machine learning in structural bioinformatics. Stat. Methods Med. Res.18, 505–526 (2009). ArticlePubMedGoogle Scholar
- Swan, A. L., Mobasheri, A., Allaway, D., Liddell, S. & Bacardit, J. Application of machine learning to proteomics data: classification and biomarker identification in postgenomics biology. OMICS17, 595–610 (2013). ArticleCASPubMedPubMed CentralGoogle Scholar
- Upstill-Goddard, R., Eccles, D., Fliege, J. & Collins, A. Machine learning approaches for the discovery of gene–gene interactions in disease data. Brief. Bioinform.14, 251–260 (2013). ArticleCASPubMedGoogle Scholar
- Yip, K. Y., Cheng, C. & Gerstein, M. Machine learning and genome annotation: a match meant to be? Genome Biol.14, 205 (2013). ArticlePubMedPubMed CentralGoogle Scholar
- Day, N., Hemmaplardh, A., Thurman, R. E., Stamatoyannopoulos, J. A. & Noble, W. S. Unsupervised segmentation of continuous genomic data. Bioinformatics23, 1424–1426 (2007). ArticleCASPubMedGoogle Scholar
- Ernst, J. & Kellis, M. ChromHMM: automating chromatin-state discovery and characterization. Nature Methods9, 215–216 (2012). This study applies an unsupervised hidden Markov model algorithm to analyse genomic assays such as ChIP–seq and DNase-seq in order to identify new classes of functional elements and new instances of existing functional element types.ArticleCASPubMedPubMed CentralGoogle Scholar
- Hoffman, M. M. et al. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nature Methods9, 473–476 (2012). ArticleCASPubMedPubMed CentralGoogle Scholar
- Chapelle, O., Schölkopf, B. & Zien, A. (eds) Semi-supervised Learning (MIT Press, 2006). BookGoogle Scholar
- Stamatoyannopoulos, J. A. Illuminating eukaryotic transcription start sites. Nature Methods7, 501–503 (2010). ArticleCASPubMedGoogle Scholar
- Boser, B. E., Guyon, I. M. & Vapnik, V. N. in A Training Algorithm for Optimal Margin Classifiers (ed. Haussler, D.) 144–152 (ACM Press, 1992). This paper was the first to describe the SVM, a type of discriminative classification algorithm.Google Scholar
- Noble, W. S. What is a support vector machine? Nature Biotech.24, 1565–1567 (2006). This paper describes a non-mathematical introduction to SVMs and their applications to life science research.ArticleCASGoogle Scholar
- Ng, A. Y. & Jordan, M. I. Advances in Neural Information Processing Systems (eds Dietterich, T. et al.) (MIT Press, 2002). Google Scholar
- Jordan, M. I. Why the logistic function? a tutorial discussion on probabilities and neural networks. Computational Cognitive Science Technical Report 9503[online], (1995). Google Scholar
- Wolpert, D. H. & Macready, W. G. No free lunch theorems for optimization. IEEE Trans. Evol. Comput.1, 67–82 (1997). This paper provides a mathematical proof that no single machine learning method can perform best on all possible learning problems.ArticleGoogle Scholar
- Yip, K. Y. et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol.13, R48 (2012). ArticleCASPubMedPubMed CentralGoogle Scholar
- Urbanowicz, R. J., Granizo-Mackenzie, D. & Moore, J. H. in Proceedings of the Parallel Problem Solving From Nature 266–275 (Springer, 2012). BookGoogle Scholar
- Brown, M. et al. in Proceedings of the Third International Conference on Intelligent Systems for Molecular Biology (ed. Rawlings, C.) 47–55 (AAAI Press, 1993). Google Scholar
- Bailey, T. L. & Elkan, C. P. in Proceedings of the Third International Conference on Intelligent Systems for Molecular Biology (eds Rawlings, C. et al.) 21–29 (AAAI Press, 1995). Google Scholar
- Schölkopf, B. & Smola, A. Learning with Kernels (MIT Press, 2002). Google Scholar
- Leslie, C. et al. (eds) Proceedings of the Pacific Symposium on Biocomputing (World Scientific, 2002). Google Scholar
- Rätsch, G. & Sonnenburg, S. in Kernel Methods in Computational Biology (eds Schölkopf, B. et al.) 277–298 (MIT Press, 2004). Google Scholar
- Zien, A. et al. Engineering support vector machine kernels that recognize translation initiation sites. Bioinformatics16, 799–807 (2000). ArticleCASPubMedGoogle Scholar
- Saigo, H., Vert, J.-P. & Akutsu, T. Optimizing amino acid substitution matrices with a local alignment kernel. BMC Bioinformatics7, 246 (2006). ArticlePubMedPubMed CentralGoogle Scholar
- Jaakkola, T. & Haussler, D. Advances in Neural Information Processing Systems 11 (Morgan Kauffmann, 1998). Google Scholar
- Shawe-Taylor, J. & Cristianini, N. Kernel Methods for Pattern Analysis (Cambridge Univ. Press, 2004). This textbook describes kernel methods, including a detailed mathematical treatment that is suitable for quantitatively inclined graduate students.BookGoogle Scholar
- Peña-Castillo, L. et al. A critical assessment of M. musculus gene function prediction using integrated genomic evidence. Genome Biol.9, S2 (2008). ArticlePubMedPubMed CentralGoogle Scholar
- Sonnhammer, E., Eddy, S. & Durbin, R. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins28, 405–420 (1997). ArticleCASPubMedGoogle Scholar
- Apweiler, R. et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res.29, 37–40 (2001). ArticleCASPubMedPubMed CentralGoogle Scholar
- Pavlidis, P., Weston, J., Cai, J. & Noble, W. S. Learning gene functional classifications from multiple data types. J. Computat. Biol.9, 401–411 (2002). ArticleCASGoogle Scholar
- Lanckriet, G. R. G., Bie, T. D., Cristianini, N., Jordan, M. I. & Noble, W. S. A statistical framework for genomic data fusion. Bioinformatics20, 2626–2635 (2004). ArticleCASPubMedGoogle Scholar
- Troyanskaya, O. G., Dolinski, K., Owen, A. B., Altman, R. B. & Botstein, D. A. Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc. Natl Acad. Sci. USA100, 8348–8353 (2003). ArticleCASPubMedGoogle Scholar
- Pearl, J. Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference (Morgan Kaufmann, 1998). This textbook on probability models for machine learning is suitable for undergraduates or graduate students.Google Scholar
- Song, L. & Crawford, G. E. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protoc.2, pdb.prot5384 (2010). Google Scholar
- Wasson, T. & Hartemink, A. J. An ensemble model of competitive multi-factor binding of the genome. Genome Res.19, 2102–2112 (2009). ArticleGoogle Scholar
- Pique-Regi, R. et al. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res.21, 447–455 (2011). ArticleCASPubMedPubMed CentralGoogle Scholar
- Cuellar-Partida, G. et al. Epigenetic priors for identifying active transcription factor binding sites. Bioinformatics28, 56–62 (2011). ArticlePubMedPubMed CentralGoogle Scholar
- Ramaswamy, S. et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl Acad. Sci. USA98, 15149–15154 (2001). ArticleCASPubMedGoogle Scholar
- Glaab, E., Bacardit, J., Garibaldi, J. M. & Krasnogor, N. Using rule-based machine learning for candidate disease gene prioritization and sample classification of cancer gene expression data. PLoS ONE7, e39932 (2012). ArticleCASPubMedPubMed CentralGoogle Scholar
- Tibshirani, R. J. Regression shrinkage and selection via the lasso. J. R. Statist. Soc. B58, 267–288 (1996). This paper was the first to describe the technique known as lasso (orL1regularization), which performs feature selection in conjunction with learning.Google Scholar
- Urbanowicz, R. J., Granizo-Mackenzie, A. & Moore, J. H. An analysis pipeline with statistical and visualization-guided knowledge discovery for Michigan-style learning classifier systems. IEEE Comput. Intell. Mag.7, 35–45 (2012). ArticlePubMedPubMed CentralGoogle Scholar
- Tikhonov, A. N. On the stability of inverse problems. Dokl. Akad. Nauk SSSR39, 195–198 (1943). This paper was the first to describe the now-ubiquitous method known asL2regularization or ridge regression.Google Scholar
- Keogh, E. & Mueen, A. Encyclopedia of Machine Learning (Springer, 2011). Google Scholar
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature489, 57–74 (2012).
- Manning, C. D. & Schütze, H. Foundations of Statistical Natural Language Processing (MIT Press, 1999). Google Scholar
- Davis, J. & Goadrich, M. Proceedings of the International Conference on Machine Learning (ACM, 2006). This paper provides a succinct introduction to precision-recall and receiver operating characteristic curves, and details under which scenarios these approaches should be used.Google Scholar
- Cohen, J. Weighted κ: nominal scale agreement provision for scaled disagreement or partial credit. Psychol. Bull.70, 213 (1968). ArticleCASPubMedGoogle Scholar
- Luengo, J., García, S. & Herrera, F. On the choice of the best imputation methods for missing values considering three groups of classification methods. Knowl. Inf. Syst.32, 77–108 (2012). ArticleGoogle Scholar
- Troyanskaya, O. et al. Missing value estimation methods for DNA microarrays. Bioinformatics17, 520–525 (2001). This study uses an imputation-based approach to handle missing values in microarray data. The method was widely used in subsequent studies to address this common problem.ArticleCASPubMedGoogle Scholar
- Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genet.46, 310–315 (2014). This study uses a machine learning approach to estimate the pathogenicity of genetic variants using a framework that takes advantage of the fact that natural selection removes deleterious variation.ArticleCASPubMedGoogle Scholar
- Qiu, J. & Noble, W. S. Predicting co-complexed protein pairs from heterogeneous data. PLoS Comput. Biol.4, e1000054 (2008). ArticlePubMedPubMed CentralGoogle Scholar
- Friedman, N., Linial, M., Nachman, I. & Pe'er, D. Using Bayesian networks to analyze expression data. J. Comput. Biol.7, 601–620 (2000). ArticleCASPubMedGoogle Scholar
- Bacardit, J. & Llorà, X. Large-scale data mining using genetics-based machine learning. Wiley Interdiscip. Rev.3, 37–61 (2013). Google Scholar
- Koski, T. J. & Noble, J. A review of Bayesian networks and structure learning. Math. Applicanda40, 51–103 (2012). Google Scholar
- Pearl, J. Causality: Models, Reasoning and Inference (Cambridge Univ. Press, 2000). Google Scholar
Author information
Authors and Affiliations
- Department of Computer Science and Engineering, University of Washington, 185 Stevens Way, Seattle, 98195–2350, Washington, USA Maxwell W. Libbrecht & William Stafford Noble
- Department of Genome Sciences, University of Washington, 3720 15th Ave NE, Seattle, 98195–5065, Washington, USA William Stafford Noble
- Maxwell W. Libbrecht
